Duchenne-Muskeldystrophie (DMD)
“Wir sind stolz darauf, der Gemeinschaft der Menschen mit seltenen Krankheiten zu dienen, und geehrt, zur weltweiten Suche nach neuen Lösungen für Duchenne beitragen zu können.”
Francesco De Santis, Vorsitzender von ITALFARMACO
Was ist Duchenne?
Die Duchenne-Muskeldystrophie ist eine seltene neuromuskuläre Erkrankung, die sich durch fortschreitende Muskelschädigung und -schwäche kennzeichnet. Aufgrund ihres X-chromosomalen Erbgangs tritt sie fast ausschließlich im männlichen Geschlecht auf. Weltweit ist schätzungsweise 1 unter 5000 männlichen Neugeborenen betroffen. Mädchen erkranken üblicherweise nicht, aber sie können Trägerinnen der genetischen Mutation sein .1,2
Duchenne Muskeldystrophie wird durch Mutationen im Dystrophin-Gen verursacht, die dazu führen, dass Dystrophin, ein wichtiges Strukturprotein, das die Muskelzellen vor Schäden infolge wiederholter Muskelkontraktionen schützt, gar nicht oder in nicht funktionsfähiger Form gebildet wird.3
Wie wirkt sich Duchenne auf den Körper aus?
Bei Menschen mit Duchenne Muskeldystrophie kann die Muskulatur durch alltägliche Aktivitäten geschädigt werden. Ein normaler, gesunder Muskel kann Schädigungen dank seiner Fähigkeit, neue Muskelzellen zu bilden, entgegenwirken. Allerdings sind die Muskeln von Menschen mit Duchenne anfälliger für Verletzungen, und ihre Fähigkeit, Muskelschäden zu reparieren, ist beeinträchtigt. Die anhaltende Schädigung der Muskelfasern führt zu chronischen Entzündungen, und die Muskelfasern werden im Verlauf der Erkankung durch Narben (Fasergewebe) und Fettablagerungen ersetzt. Im Laufe der Zeit verlieren die Muskeln ihre Fähigkeit zur Reperatur und ihre Fähigkeit, sich zusammenzuziehen, was zu einem Verlust an Muskelmasse und zu Muskelschwäche führt. 4 – 7
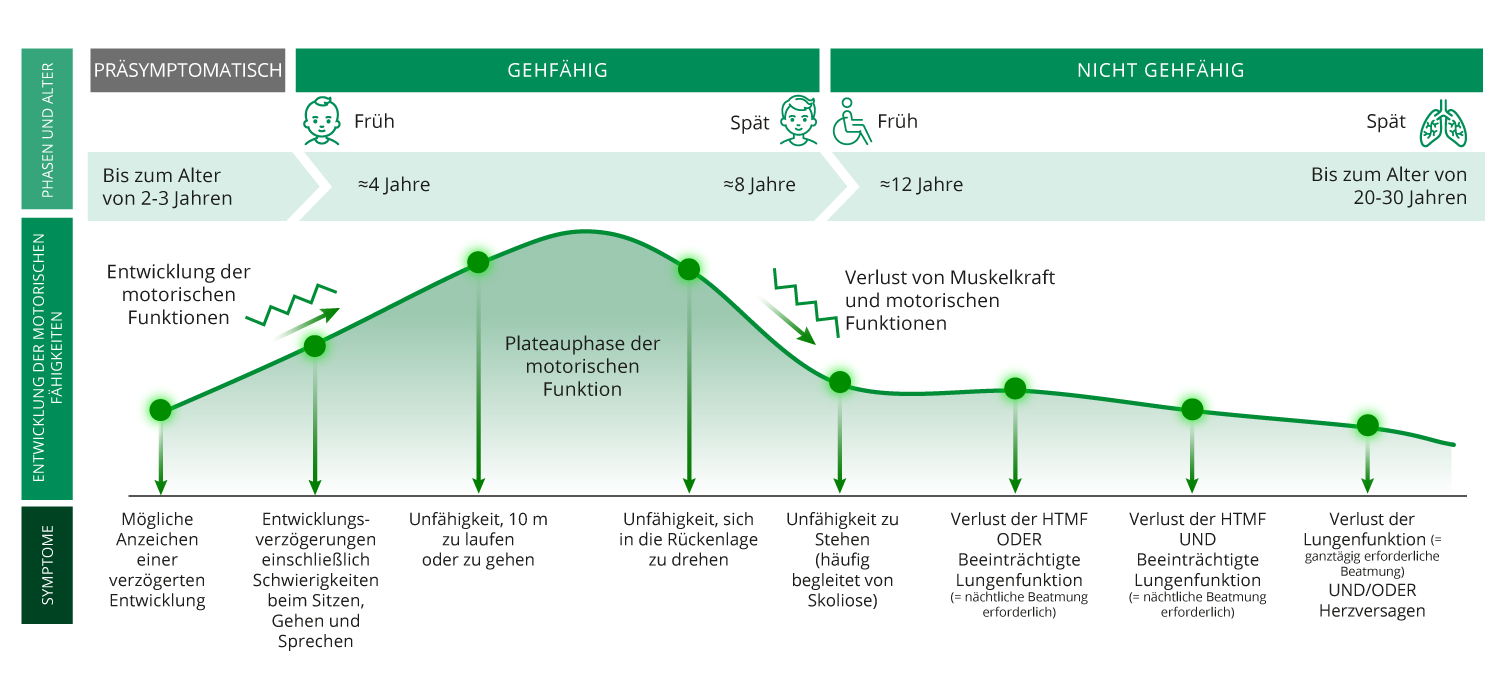
DMD: Duchenne Muskel Dystrophie; HTMF: Hand-to-Mouth-Function.
1. Broomfield J, Hill M, Guglieri M, Crowther M, Abrams K. Life Expectancy in Duchenne Muscular Dystrophy: Reproduced Individual Patient Data Meta-analysis. Neurology. 2021;97(23):e2304-e2314.
2. Landfeldt E, Lindgren P, Bell CF, et al. The burden of Duchenne muscular dystrophy: an international, cross-sectional study. Neurology. 2014;83(6):529-536.
3. Muntoni F, Signorovitch J, Sajeev G, et al. DMD Genotypes and Motor Function in Duchenne Muscular Dystrophy: A Multi-institution Meta-analysis With Implications for Clinical Trials. Neurology. 2023;100(15):e1540-e1554.
Leben mit Duchenne
Überall auf der Welt, in Europa, in den Vereinigten Staaten und anderenorts, erzählen Menschen aus der Duchenne-Gemeinschaft Geschichten von Stärke und Zuversicht:
“Es ist gut, zu wissen, dass die meisten Duchenne-Kinder glückliche Kinder sind und die meisten Familien nach dem ersten Schock der Diagnose mit ihr sehr gut umgehen können.”
Elizabeth Vroom, World Duchenne Organization (UPPMD) – Diagnose und Behandlung der Duchenne-Muskeldystrophie – Ein Leitfaden für Familien
“Nachdem wir eine gesicherte Diagnose erhalten hatten, konnten wir unseren Kampf endlich in Begriffe fassen. Es gibt immer Hoffnung, weil wir Tag für Tag unermüdlich auf eine Zukunft ohne Duchenne hinarbeiten.”
Silvia Avila, Vorsitzende des Duchenne Parent Project España (DPPE) , Spanien
“Als das Wort Duchenne vor 10 Jahren in unser Leben einbrach, war das ein Schock – ich konnte es zuerst nicht einmal aussprechen. Langsam wurde uns klar, dass wir einander beistehen können, dass es in dieser Gemeinsamkeit Wege nach vorne gibt, dank einer Gemeinschaft von Familien, die bereit sind, Erfahrungen auszutauschen, und einer internationalen wissenschaftlichen Gemeinschaft, deren Studien Hoffnung geben.”
Mutter eines Duchenne-Kindes, Parent Project Italia
Klinische
Arzneimittelstudien
Nützliche Links
Die Gemeinschaft der Menschen mit seltenen Krankheiten einschließlich von Duchenne Muskeldystrophie konzentriert sich darauf, das Bewusstsein für diese Krankheit zu schärfen und den Kontakt zwischen den Familien zu erleichtern, indem sie Ressourcen für Aufklärung, Forschung und Unterstützungsdienste bereitstellt. Hier finden Sie einige von vielen regionalen Patienten- und Wohltätigkeitsorganisationen, bei denen Sie weitere nützliche Informationen und Verbindungen finden können.
Verein Marathon
Der Verein Marathon ist eine österreichische Patientenorganisation, die sich für Menschen mit Duchenne-Muskeldystrophie und anderen neuromuskulären Erkrankungen einsetzt. Ziel des Vereins ist es, Betroffene und ihre Familien bestmöglich zu unterstützen, sei es durch Information, individuelle Beratung, Vernetzung oder den Einsatz für eine bestmögliche medizinische Versorgung und gesellschaftliche Teilhabe.
Ein besonderer Fokus liegt auf der Förderung der Transition vom Kindes- ins Erwachsenenalter sowie auf der Zusammenarbeit mit medizinischen Fachkräften und Forschungseinrichtungen.
Bitte beachten Sie, dass die Auflistung der Patientenorganisationen möglicherweise nicht vollständig ist.